Base STO-nG
Une base STO-nG est une base utilisée en chimie numérique, c'est-à-dire un ensemble de fonctions utilisées pour créer des orbitales moléculaires.

Catégories :
Chimie quantique - Physique quantique - Chimie numérique
Recherche sur Google Images :

Source image : fr.wikipedia.org Cette image est un résultat de recherche de Google Image. Elle est peut-être réduite par rapport à l'originale et/ou protégée par des droits d'auteur. |
Page(s) en rapport avec ce sujet :
- Le dispositif de base STON-31G, mais non le STO - NG, donne des déplacements énergétiques Epv... base ; Orbitale gaussienne ; Méthode ab initio ; Niveau énergie... (source : cat.inist)
- liées aux surfaces d'énergie potentielle et théorie des orbitales frontières...... L'usage est fait aujourd'hui des bases suivantes : STO - nG (n = 2-6)... (source : www1.univ-batna)
Une base STO-nG est une base utilisée en chimie numérique, c'est-à-dire un ensemble de fonctions utilisées pour créer des orbitales moléculaires. Plus exactement, une base STO-nG est une base minimale, dans laquelle la lettre n sert à désigner le nombre de fonctions primitives gaussiennes que comprend une base simple. Pour les bases minimales, les orbitales de cœur et de valence sont représentées par le même nombre de fonctions primitives gaussiennes Φi. Ainsi, une base STO-3G pour la description de l'orbitale 1s de l'atome d'hydrogène est une combinaison linéaire de 3 fonctions primitives gaussiennes. Il est aisé de calculer l'énergie d'un électron de l'orbitale 1s de l'atome d'hydrogène représenté dans les bases STO-nG.
Dans les sections suivantes, la structure des bases minimales STO-nG sont expliquées en se basant sur l'exemple de l'atome H.
Description
Base STO-1G
Ici, ψ (1sH) =ψ (STO-1G) =c1Φ1 dans laquelle c1=1 et 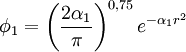 . La valeur optimale de α1 est celle qui donne la valeur minimale de l'énergie pour l'électron 1s de l'atome d'hydrogène. L'exposant α1 pour la base STO-1G peut être déduit manuellement en dérivant l'énergie comparé à l'exposant et en prenant sa valeur en 0.
. La valeur optimale de α1 est celle qui donne la valeur minimale de l'énergie pour l'électron 1s de l'atome d'hydrogène. L'exposant α1 pour la base STO-1G peut être déduit manuellement en dérivant l'énergie comparé à l'exposant et en prenant sa valeur en 0.
La valeur de α1 est donc : 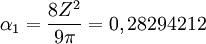 et , pour cette valeur, l'énergie d'un électron 1s d'un atome H peut être évaluée à -0, 42441318 hartree. L'expression de l'énergie de l'électron 1s de l'atome H est fonction uniquement de c1 et d'autres constantes principales comme π. Par convenance, les détails de la base sont représentés comme ci-dessous :
et , pour cette valeur, l'énergie d'un électron 1s d'un atome H peut être évaluée à -0, 42441318 hartree. L'expression de l'énergie de l'électron 1s de l'atome H est fonction uniquement de c1 et d'autres constantes principales comme π. Par convenance, les détails de la base sont représentés comme ci-dessous :
| STO-1G |  |
 |
| 0, 2829421200 | 1, 0000000000 |
Base STO-2G
En général, une base STO-nG est une combinaison linéaire de n fonctions gaussiennes primitives. Les bases STO-nG sont généralement représentées par leurs exposants et les cœfficients correspondants. Ainsi, un base STO-2G[1] qui est une combinaison linéaire de deux fonctions primitives gaussiennes peut être représentée comme ci-dessous :
| STO-2G | |
|
| 0, 1309756377×101 | 0, 4301284983 | |
| 0, 2331359749 | 0, 6789135305 |
Calcul de l'énergie électronique dans les bases STO-nG
L'énergie électronique d'un dispositif est calculée comme la valeur attendue du hamiltonien électronique :
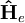 est le hamiltonien électronique du dispositif. Les valeurs attendues peuvent être analytiquement obtenues uniquement pour un dispositif à deux corps, comme un atome d'hydrogène par exemple. Le hamiltonien pour un atome d'hydrogène est donné par :
est le hamiltonien électronique du dispositif. Les valeurs attendues peuvent être analytiquement obtenues uniquement pour un dispositif à deux corps, comme un atome d'hydrogène par exemple. Le hamiltonien pour un atome d'hydrogène est donné par :
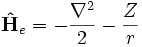 .
.
Les intégrales exactes pour l'énergie cinétique, les valeurs attendues de l'énergie potentielle et les intégrales de recouvrement peuvent être obtenues comme indiqué ci-après :
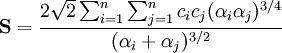 .
.
Ainsi, lors de l'utilisation d'une base STO-nG avec n gaussiennes primitives, il y a n2 intégrales d'énergie cinétique, n2 intégrales d'énergie potentielle et n2 intégrales de recouvrement. Ainsi, avec n fonctions primitives gaussiennes dans la base, on a besoin de calculer 3n2 intégrales.
Annexe
Les bases STO-nG (pour n=2, 3 et 6) peuvent être consultées depuis la banque de données en ligne[2] et l'énergie de l'électron 1s de l'atome H peut aisément être calculée à la main ou en utilisant un programme simple. Ci-dessous est proposé un programme en Fortran 77 dans laquelle l'expression de l'énergie est explicitement donnée et en donnant la base comme donnée d'entrée, l'énergie est obtenue en sortie.
!---------------------------------------------------------------- ! PROGRAM sto_ng CALCULATES THE ENERGY OF 1s ELECTRON OF "H" ATOM ! OR OTHER HYDROGENIC ATOMIC SYSTEMS WITH MINIMAL BASIS SETS. THE ! PROGRAM CAN BE EASILY EXTENDED FOR LARGER BASIS SETS. !---------------------------------------------------------------- PROGRAM sto_ng IMPLICIT NONE !---------------------------------------------------------------- ! i AND j : DUMMY INDICES ! n : NUMBER OF PRIMITIVE GTOs ! Z : ATOMIC NUMBER !---------------------------------------------------------------- INTEGER i, j, n, Z !---------------------------------------------------------------- ! V(i,j) : i,j TH ELEMENT OF THE POTENTIAL ENERGY MATRIX ! T(i,j) : i,j TH ELEMENT OF THE KINETIC ENERGY MATRIX ! S(i,j) : i,j TH ELEMENT OF THE OVERLAP INTEGRAL MATRIX ! VI : TOTAL SUM OF ALL POTENTIAL ENERGY INTEGRALS ! TI : TOTAL SUM OF ALL KINETIC ENERGY INTEGRALS ! SI : TOTAL SUM ALL OF OVERLAP INTEGRALS ! c(i) : i TH COEFFICIENT !alpha(i) : i TH EXPONENT !---------------------------------------------------------------- DOUBLE PRECISION V(100,100), T(100,100), S(100,100) DOUBLE PRECISION alpha(100), c(100), VI, TI, SI, PI PI=3.1415926535898D0 OPEN(UNIT=1, FILE="input.txt") OPEN(UNIT=2, FILE="output.txt") READ(1,*)Z,n DO i=1,n READ(1,*)alpha(i),c(i) ENDDO !---------------------------------------------------------------- ! CALCULATION OF OVERLAP INTEGRALS AND THEIR SUMMATION !---------------------------------------------------------------- DO i=1,n DO j=1,n S(i,j)=c(i)*c(j)*2.0D0*SQRT(2.0D0)*(alpha(i)*alpha(j))**0.75D &0/(alpha(i)+alpha(j))**(1.5D0) ENDDO ENDDO SI=0.0D0 DO i=1,n DO j=1,n SI=SI+S(i,j) ENDDO ENDDO !---------------------------------------------------------------- ! CALCULATION OF KINETIC ENERGY INTEGRALS AND THEIR SUMMATION !---------------------------------------------------------------- DO i=1,n DO j=1,n T(i,j)=c(i)*c(j)*6.0D0*SQRT(2.0D0)*(alpha(i)*alpha(j))**1.75D0/ &(alpha(i)+alpha(j))**(2.5D0) ENDDO ENDDO TI=0.0D0 DO i=1,n DO j=1,n TI=TI+T(i,j) ENDDO ENDDO !---------------------------------------------------------------- ! CALCULATION OF POTENTIAL ENERGY INTEGRALS AND THEIR SUMMATION !---------------------------------------------------------------- DO i=1,n DO j=1,n V(i,j)=-c(i)*c(j)*4.0D0*SQRT(2.0D0)*Z*(alpha(i)*alpha(j))**0. &75D0/(SQRT(PI)*(alpha(i)+alpha(j))) ENDDO ENDDO VI=0.0D0 DO i=1,n DO j=1,n VI=VI+V(i,j) ENDDO ENDDO WRITE(2,*)"\n\nBasis set :\n" WRITE(2,002)"ALPHA(i)","C(i)" DO i=1,n WRITE(2,003)alpha(i),c(i) ENDDO WRITE(2,001)"\n\nK.E. integral is :", TI," hartree" WRITE(2,001)"\nP.E. integral is :", VI," hartree" WRITE(2,001)"\nOverlap Integral is :", SI," hartree" WRITE(2,001)"\nEnergy of H atom is :", (VI+TI)/SI," hartree/partic &le" WRITE(2,001)"\nENERGY of H atom is :",(VI+TI)*27.211397D0/SI," e.V &./particle" WRITE(2,001)"\nENERGY of H atom is :",(VI+TI)*627.509D0/SI," kcal/ &mol" WRITE(2,001)"\nENERGY of H atom is :",(VI+TI)*2625.51D0/SI," kJ/mo &l" WRITE(2,001)"\nENERGY of H atom is :",(VI+TI)*219475D0/SI," cm-1" 001 FORMAT(A,D20.10,A) 002 FORMAT(8X,A,19X,A) 003 FORMAT(D20.10,6X,D20.10) 004 FORMAT(D20.10) CLOSE(1) CLOSE(2) STOP ENDINPUT FILE DETAILS FILE : input.txt 1 ! ATOMIC NUMBER 2 ! NO. OF PRIMITIVE GTOs 0.1309756377D+01 0.4301284983D+00 ! BASIS SET alpha c 0.2331359749D+00 0.6789135305D+00OUTPUT FILE DETAILS FILE : output.txt Basis set : ALPHA(i) C(i) 0.1309756377E+01 0.4301284983E+00 0.2331359749E+00 0.6789135305E+00 K.E. integral is : 0.7348827001E+00 hartree P.E. integral is : -0.1189280102E+01 hartree Overlap Integral is : 0.1000000000E+01 hartree Energy of H atom is : -0.4543974016E+00 hartree/particle ENERGY of H atom is : -0.1236478809E+02 e.V./particle ENERGY of H atom is : -0.2851384591E+03 kcal/mol ENERGY of H atom is : -0.1193024922E+04 kJ/mol ENERGY of H atom is : -0.9972886973E+05 cm-1Références
- ↑ (en) EMSL Basis Set Exchange
- ↑ Erreur de citation : Balise
<ref>incorrecte ; aucun texte n'a été apporté pour les références appeléespln.
- (en) Cet article est partiellement ou en totalité issu d'une traduction de l'article de Wikipédia en anglais intitulé «STO-nG basis sets».
Recherche sur Amazone (livres) : |
Voir la liste des contributeurs.
La version présentée ici à été extraite depuis cette source le 12/04/2009.
Ce texte est disponible sous les termes de la licence de documentation libre GNU (GFDL).
La liste des définitions proposées en tête de page est une sélection parmi les résultats obtenus à l'aide de la commande "define:" de Google.
Cette page fait partie du projet Wikibis.
 Accueil
Accueil Recherche
Recherche Début page
Début page Contact
Contact Imprimer
Imprimer Accessibilité
Accessibilité